Natural language generation has improved remarkably over the last decade and OpenAI’s ChatGPT showcases an unbelievable performance of state-of-the art systems. Like many curious people over the last month, I played with ChatGPT from time to time. It’s super impressive – it can create poems, write prose in nearly any style, and provide detailed answers to all sorts of questions. Oh, and it’s also an amazing bullshit artist and it will lie with abandon.
I decided to ask ChatGPT a specific question about particle physics that is a bit subtle but has a straightforward answer. ChatGPT’s response is stunning bullshit.

ChatGPT gets the first part right – the most common decay mode is to an electron, an electron antineutrino, and a muon neutrino. Now, the rest of the answer is completely wrong. First, the muon is kinematically forbidden from decaying into pions. The muon is simply not massive enough to do so. Second, even if the muon were massive enough to decay with three charged pions in the final state (like the tau lepton can), conservation of lepton number still requires neutrinos. Apparently ChatGPT has thrown lepton number overboard so that it can give me an answer!
The correct answer to my quetsion is: Theoretically yes, but hopelessly rare. No observation of muon decay without emitting neutrinos has ever been observed.
In the Standard Model (the theory of fundamental particle interactions) with massless neutrinos, muon decay must include neutrinos in the final state. The Standard Model with massless neutrinos conserves family lepton number and so neutrinos in the final state are required to conserve muon and electron number (a muon neutrino and an electron antineutrino). However, neutrino flavour oscillation observations tell us that neutrinos have very, very tiny masses. Extending the Standard Model to include neutrino masses allows for the muon to decay without neutrinos (for example to an electron and a photon) but at rates so extremely small that we have no hope of observing neutrinoless decays of the muon unless the Standard Model is further extended with exotic physics.
Ok, rather technical stuff. But where did ChatGPT come up with the idea of pions in the final state? Well, I asked it. And sure enough ChatGPT came up with references, including a paper in Nature from 1982 with claims of actually observing neutrinoless decays of the muon! Well, either I missed something during my PhD and postdocs (I have done a fair amount of work on supersymmetric extensions of the Standard Model with enhanced flavour violating decays of the muon) or ChatGPT is making stuff up. All of the references that ChaptGPT gave me are complete fabrications – they don’t exist!!

It turns out that natural language generation in systems like ChatGPT suffer from hallucinating unintended text. Its creative power, its ability to write poems etc., requires such an enormous amount of flexibility that ChatGPT seems to start making things up to complete an answer. I’m not an expert on natural language processing, but I know that researchers are aware of the phenomenon.
I happen to know something about particle physics so it’s not a big deal that ChatGPT fibbed to answer my question. In fairness, my question is a bit subtle (short answer is No with an if, long answer is Yes with a but). But imagine circumstances that are more important. How do you know if you can trust the answers? Asking it for reference doesn’t seem to help.
To me the lesson is that systems like ChatGPT are going to be ever more useful with the potential to make life much more convenient. But these systems are not a substitute for human cognition. For the teachers out there – don’t worry about ChatGPT writing your students’ essays, just check to see if the references exist!
Update (February 23, 2023)
I returned to ChatGPT last night, getting into a discussion with it about the top quark. ChatGPT continued to give insane answers about QCD, electroweak physics, and the nature of the Higgs sector. I thought I could get it on track if it would just realize that the muon’s mass is less than the pions. It produced this gem:

ChatGPT fails the Piaget test.


 , given by the convolution with a negative sum, namely,
, given by the convolution with a negative sum, namely,
 has support on
has support on  , and
, and  is the Poisson. The resulting distribution can be expressed in terms of the modified Bessel function,
is the Poisson. The resulting distribution can be expressed in terms of the modified Bessel function,


 is the arrival rate and
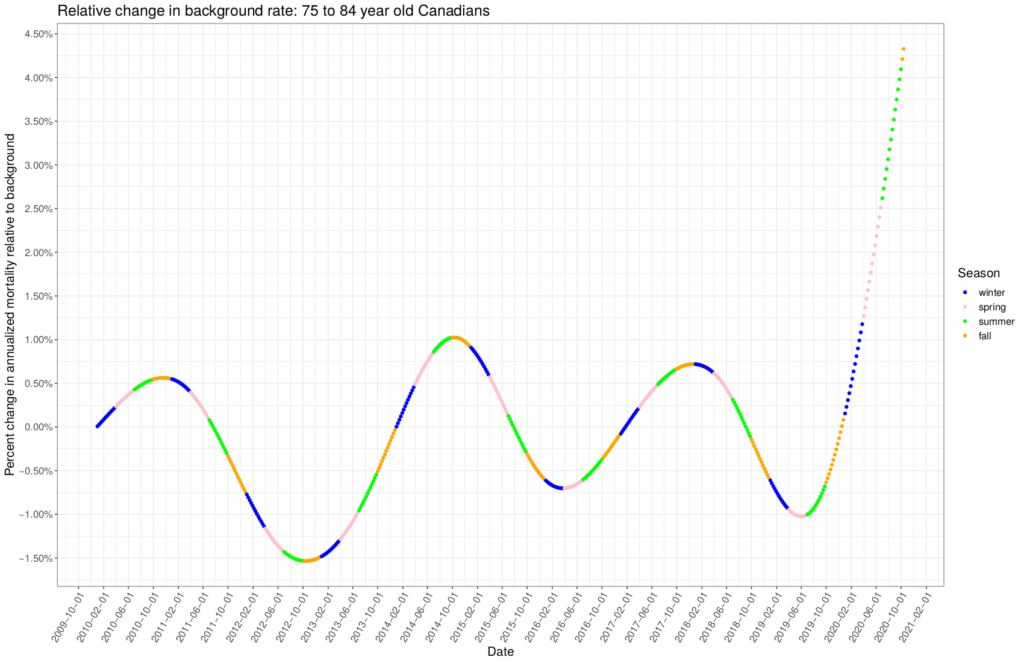
is the arrival rate and  is the average service rate. For example, given an arrival rate of 50 people per day and an average length of stay of 14 days in the ICU, the steady-state average number of people in the ICU would be 700 patients, provided the number of beds (servers) is larger than 700. I don’t know if the length of stay has been varying over time. It would be great to do a full time dependent competing risk survival analysis on that data, but the steady rise in the departure rate that we see over the winter might indicate that the length of stay in the ICU has also been changing with the arrival rate. I don’t know.
is the average service rate. For example, given an arrival rate of 50 people per day and an average length of stay of 14 days in the ICU, the steady-state average number of people in the ICU would be 700 patients, provided the number of beds (servers) is larger than 700. I don’t know if the length of stay has been varying over time. It would be great to do a full time dependent competing risk survival analysis on that data, but the steady rise in the departure rate that we see over the winter might indicate that the length of stay in the ICU has also been changing with the arrival rate. I don’t know.
 , and a trend spline,
, and a trend spline,  ,
, 









 , as a compound Poisson process,
, as a compound Poisson process,
 is the number of infection events that arrived up to time
is the number of infection events that arrived up to time  , and
, and  is the number infected at each infection event. We model
is the number infected at each infection event. We model 
 . The infection events arrive exponentially distributed in time with arrival rate
. The infection events arrive exponentially distributed in time with arrival rate  . The characteristic function for
. The characteristic function for ![\begin{align*}\phi_{Q(t)}(u) &=\mathbb{E}[e^{iuQ(t)}] \\ &= \exp\left(rt\ln\left(\frac{1-p}{1-pe^{iu}}\right)\right) \\ &= \left(\frac{1-p}{1-pe^{iu}}\right)^{rt},\end{align*}](https://maybury.ca/the-reformed-physicist/wp-content/ql-cache/quicklatex.com-493e9b3cc164dbdfe835dadda287c3ca_l3.png "Rendered by QuickLaTeX.com")
 and thus
and thus 
 . The infection events occur continuously in time according to the Poisson arrivals. However, the communicable period,
. The infection events occur continuously in time according to the Poisson arrivals. However, the communicable period,  , which we model as a gamma process,
, which we model as a gamma process,
 . By promoting the communicable period to a random variable, the negative binomial process changes into a Levy process with characteristic function,
. By promoting the communicable period to a random variable, the negative binomial process changes into a Levy process with characteristic function,![\begin{align*}\mathbb{E}[e^{iuZ(t)}] &= \exp(-t\psi(-\eta(u))) \\ &= \left(1- \frac{r}{b}\ln\left(\frac{1-p}{1-pe^{iu}}\right)\right)^{-at},\end{align*}](https://maybury.ca/the-reformed-physicist/wp-content/ql-cache/quicklatex.com-b8dec93aa84eef4c5279cb4cf8a9011b_l3.png "Rendered by QuickLaTeX.com")
 , the Levy symbol, and
, the Levy symbol, and  , the Laplace exponent, are respectively given by,
, the Laplace exponent, are respectively given by,![\begin{align*}\mathbb{E}[e^{iuQ(t)}] &= \exp(t\,\eta(u)) \\\mathbb{E}[e^{-sT(t)}] &= \exp(-t\,\psi(s)), \end{align*}](https://maybury.ca/the-reformed-physicist/wp-content/ql-cache/quicklatex.com-a388ec11ca6d71673b206a186378daf0_l3.png "Rendered by QuickLaTeX.com")

 is the random number of people infected by a single infected individual over her random communicable period and is further over-dispersed relative to a pure negative binomial process, getting us closer to driving propagation through super-spreader events . The characteristic function in eq.(6) for the number of infections from a single infected person gives us the entire model. The basic reproduction number
is the random number of people infected by a single infected individual over her random communicable period and is further over-dispersed relative to a pure negative binomial process, getting us closer to driving propagation through super-spreader events . The characteristic function in eq.(6) for the number of infections from a single infected person gives us the entire model. The basic reproduction number  is,
is,
 , such as the number of infectious individuals at time
, such as the number of infectious individuals at time  where
where  is the random communicable period) the expectation of the process follows,
is the random communicable period) the expectation of the process follows,
 is the counting process (
is the counting process (

 is called the Malthusian parameter and it controls the exponential growth of the process. Because the renewal equation gives us eq.(11), we can build a Bayesian hierarchical model for inference with just cumulative count data. We take US county data,
is called the Malthusian parameter and it controls the exponential growth of the process. Because the renewal equation gives us eq.(11), we can build a Bayesian hierarchical model for inference with just cumulative count data. We take US county data,  -effective across the United States. We use
-effective across the United States. We use 
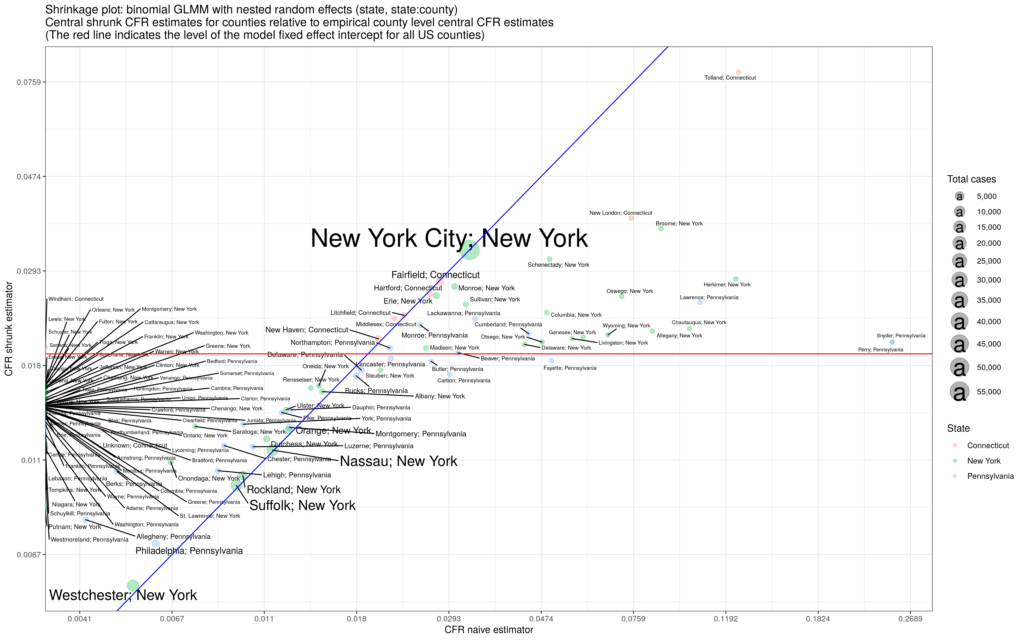
 is the county label; the variance parameters use half-Cauchy priors and the fixed and random effects use normal priors. We estimate the model and generate posterior distributions for all parameters. The result for the United States using data over the summer is the figure below:
is the county label; the variance parameters use half-Cauchy priors and the fixed and random effects use normal priors. We estimate the model and generate posterior distributions for all parameters. The result for the United States using data over the summer is the figure below:
 across the US singles out the Midwestern states and Hawaii as hot-spots while Arizona sees no county with exponential growth.
across the US singles out the Midwestern states and Hawaii as hot-spots while Arizona sees no county with exponential growth.  observations taking the form of a 6-tuple:
observations taking the form of a 6-tuple:
 : index of parent,
: index of parent,  : time of birth,
: time of birth,  : time of death,
: time of death,  : number of offspring birth events,
: number of offspring birth events,  : number of offspring.
: number of offspring.

 are hyper-parameters.
are hyper-parameters.